
阿片受体激动剂
阿片镇痛药物: 军科院的遗憾和扬子江的奇迹
2017-10-16 22:52:29
来自:医药研发社交平台
作者:雨里
阅读量:1
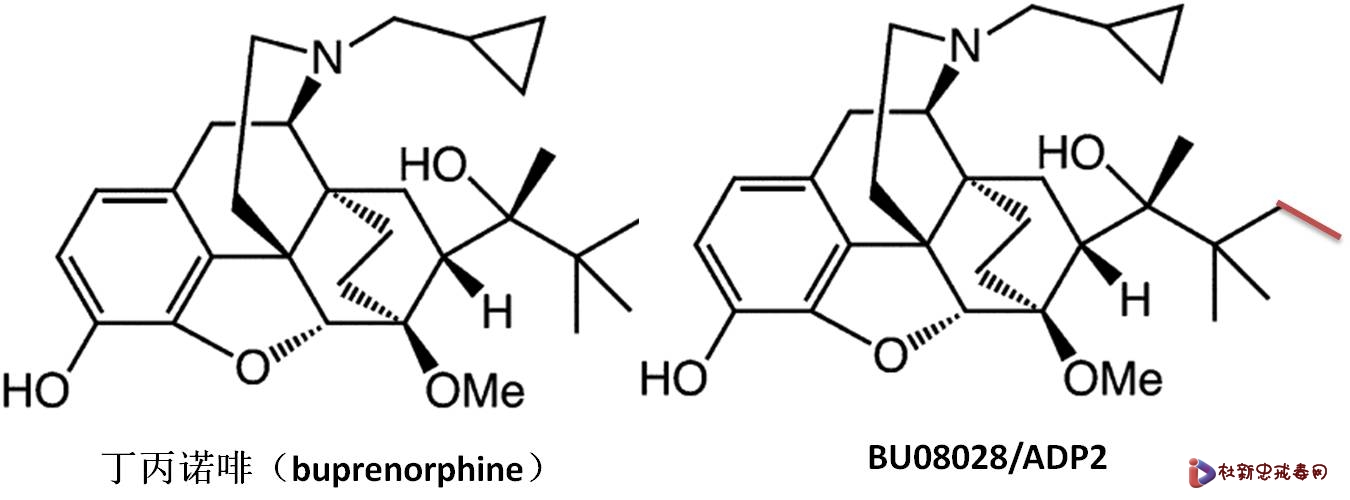
最近美国科学院院报(PNAS)发表了一篇阿片镇痛药物的文章:英国Bath大学的科学家在猴子试验中发现丁丙诺啡(buprenorphine)的类似物BU08028,虽然结构上只比丁丙诺啡侧链上多一个甲基,却具有独特的药理活性(细节见后面分析),不但镇痛活性高于丁丙诺啡,而且克服了阿片类药物常见的副作用和成瘾性(1)。美国Buffalo大学的李俊旭教授在评论文章中给予这一发现高度评价,文章的标题就是“离阿片研究的圣杯(Holy Grail)又近一步”(2)。军科院的仲伯华教授看到这篇文章后却发现,化合物BU08028就是他实验室90年代设计合成的化合物ADP2,该化合物在一系列药理实验中显示了优于丁丙诺啡的活性。当时并没有发表文章,但化合物的合成及药理研究包含在研究生吴波1999年的硕士论文之中(3)。 事实上,在秦伯益院士和仲伯华教授的领导下,军科院药物研究所的研究团队从90年代初即开始了阿片镇痛药物的研发,发现了一系列具有极好活性的候选化合物。其中,噻吩诺啡作为戒毒药物,已进入临床研究。然而,目前来看,最具特色的ADP2,也就是PNAS发表的BU08028,却错过了开发的最佳时机。这不能不说是一个遗憾。
说到阿片类麻醉或镇痛药物,作者本人2009年在扬子江工作时,见证了扬子江的重磅炸弹地佐辛的上市。地佐辛是1970年合成的阿片μ受体的部分激动剂,由Wyeth于1986年开发上市,用于手术镇痛。2000年被当时的制造商阿斯利康自愿撤出市场,不再生产。估计有阿斯利康战略布局的问题,但主要还是效果和副作用与吗啡等其他阿片类药物相比,没有太大优势。虽然药物滥用的趋势比吗啡小,但仍然受到管控。然而,扬子江在2009年将其在中国按3.1类新药开发,独家上市后,迅速占据国内镇痛药物市场。2016年,年销售超过40亿,占到国内阿片类药物市场份额的44%。这不能不说是一个奇迹,不知当时自愿选择退市的阿斯利康做何感想。

军科院的遗憾和扬子江的奇迹,折射出中国当前新药研发面临的居多问题,既有政策方面的,也有科学和思维方式方面的,令人思考。本文结合阿片类药物的历史和当前研发策略分享个人的一些看法。
正 文
阿片类药物的回顾
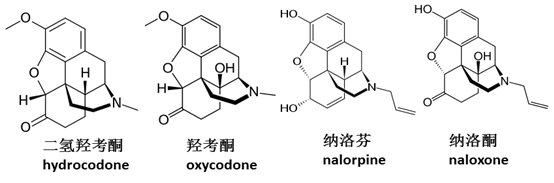
疼痛(pain)是人们去医院就医的最主要起始原因。影响的病人超过糖尿病、心血管和肿瘤病人之和。疼痛的原因和形式各种各样,几乎伴随所有疾病,而且发病机制不一。既有物理的受伤,也有原因不明的神经疼痛。许多疼痛,目前临床上无药可治。比如截肢病人常见的“幻肢痛”,也就是感觉到并不存在的肢体疼痛,中西医都束手无策。 阿片类药物是临床上最常见和最有效的镇痛药物。阿片其实是鸦片的谐音。没有人准确知道什么时候人类开始使用鸦片。最早有记载的鸦片种植和分离的是公元前第三世纪,居住在当今伊拉克境内的古苏美尔人。公元8世纪,鸦片从阿拉伯传入中国。鸦片常用于宗教仪式,代表可以带走病痛的上帝之物(4)。1806年,Sertuner等人从鸦片中分离主要活性成分吗啡。因为有梦境一般的镇痛效果,吗啡morphine实际上是取名于睡神morpheus。1850年注射器应用于临床后,吗啡作为镇痛和麻醉药物得到广泛应用。不幸的是,吗啡既是天使又是魔鬼。随着大规模应用,其魔鬼的一面开始展现,副作用包括便秘、抑制呼吸系统等。过量时,这些副作用可以是致命的。而这还不是重点,最可怕的是成瘾性。可以说吗啡打开了毒品这个潘多拉魔盒,从海洛因到新型的苯丙胺类毒品,都带来了巨大的社会问题。毒品的厉害,大家都有所耳闻,它可以将一个人变成魔鬼。瘾君子为获得毒品,不择手段,六亲不认。这种成瘾性不是人类所能抗拒的,因为毒品改变了大脑的神经回路,身不由己。我在哥伦比亚医学院时,看过成瘾性研究的动物实验。老鼠笼子里,一边是正常的食物,一边是混有毒品的食物。老鼠上瘾后,将有毒品的隔间关门上锁。老鼠会用头撞击上锁的门,直至头破血流而死。在此奉劝所有人远离毒品,一旦沾上,家破人亡。 因此,人类从19世纪初开始,拉开了寻找高效低毒、成瘾性小的阿片类药物的征程。1898年,乙酰化的吗啡被合成,活性高于吗啡,但成瘾性也更高,这就是着名的毒品海洛因。1939和1946年,分别发现骨架简化的阿片药物杜冷丁和美沙酮。无一例外,这些药物都具有类似于吗啡的镇痛活性和成瘾性。以致于意外发现杜冷丁的Schaumann得出结论,不可能发现具有吗啡镇痛活性但没有成瘾性的物质。后来开发的二氢羟考酮和羟考酮,宣称副作用更小,但相比于吗啡,并没有革命性的改善。
疼痛(pain)是人们去医院就医的最主要起始原因。影响的病人超过糖尿病、心血管和肿瘤病人之和。疼痛的原因和形式各种各样,几乎伴随所有疾病,而且发病机制不一。既有物理的受伤,也有原因不明的神经疼痛。许多疼痛,目前临床上无药可治。比如截肢病人常见的“幻肢痛”,也就是感觉到并不存在的肢体疼痛,中西医都束手无策。 阿片类药物是临床上最常见和最有效的镇痛药物。阿片其实是鸦片的谐音。没有人准确知道什么时候人类开始使用鸦片。最早有记载的鸦片种植和分离的是公元前第三世纪,居住在当今伊拉克境内的古苏美尔人。公元8世纪,鸦片从阿拉伯传入中国。鸦片常用于宗教仪式,代表可以带走病痛的上帝之物(4)。1806年,Sertuner等人从鸦片中分离主要活性成分吗啡。因为有梦境一般的镇痛效果,吗啡morphine实际上是取名于睡神morpheus。1850年注射器应用于临床后,吗啡作为镇痛和麻醉药物得到广泛应用。不幸的是,吗啡既是天使又是魔鬼。随着大规模应用,其魔鬼的一面开始展现,副作用包括便秘、抑制呼吸系统等。过量时,这些副作用可以是致命的。而这还不是重点,最可怕的是成瘾性。可以说吗啡打开了毒品这个潘多拉魔盒,从海洛因到新型的苯丙胺类毒品,都带来了巨大的社会问题。毒品的厉害,大家都有所耳闻,它可以将一个人变成魔鬼。瘾君子为获得毒品,不择手段,六亲不认。这种成瘾性不是人类所能抗拒的,因为毒品改变了大脑的神经回路,身不由己。我在哥伦比亚医学院时,看过成瘾性研究的动物实验。老鼠笼子里,一边是正常的食物,一边是混有毒品的食物。老鼠上瘾后,将有毒品的隔间关门上锁。老鼠会用头撞击上锁的门,直至头破血流而死。在此奉劝所有人远离毒品,一旦沾上,家破人亡。 因此,人类从19世纪初开始,拉开了寻找高效低毒、成瘾性小的阿片类药物的征程。1898年,乙酰化的吗啡被合成,活性高于吗啡,但成瘾性也更高,这就是着名的毒品海洛因。1939和1946年,分别发现骨架简化的阿片药物杜冷丁和美沙酮。无一例外,这些药物都具有类似于吗啡的镇痛活性和成瘾性。以致于意外发现杜冷丁的Schaumann得出结论,不可能发现具有吗啡镇痛活性但没有成瘾性的物质。后来开发的二氢羟考酮和羟考酮,宣称副作用更小,但相比于吗啡,并没有革命性的改善。

1942年,第一个阿片拮抗剂纳洛芬(nalorpine)被发现,可以抵消吗啡的部分作用。随后,安全性更好的拮抗剂药物纳洛酮(naloxone)开发上市,用于治疗吗啡类镇痛药物的副作用,戒断反应和戒毒。
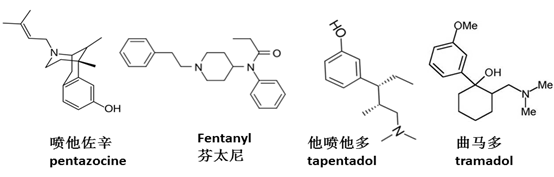
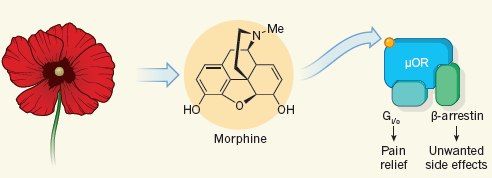
60年代中期,阿片类药物的作用机制研究逐渐深入。这些药物的镇痛活性、副作用以及成瘾性,被认为是通过体内的阿片受体调节的。阿片受体有不同的亚型,包括μ,κ,δ受体,以及后来发现的ORL(opiate receptor like)或NOP受体。1975年,Kosterlitz等首次发现阿片受体内源性配体(enkephalin)。随后,多种内源性肽类配体被发现,调节不同的生理功能。上述化学药物,具有共同的结构特征,一个质子化的N原子,平行处于芳香环的上方,这一特征使得这些药物与阿片受体有高度亲合性,起到激动或拮抗作用。 作用机制的研究导致更多具有独特药理活性的药物被发现。吗啡是μ受体的激动剂。1960年,Jassen的研发人员发现4-anilinopiperidines结构可以获得高活性的μ受体的激动剂,导致最强效的阿片类药物芬太尼及其类似物的问世。由于芬太尼的半衰期很短,活性高,是理想的手术镇痛药物。70年代发现κ受体的激动剂喷他佐辛(pentazocine),该药物具有较低的成瘾性。有证据表明,κ受体的选择性激动剂,可能减少成瘾性。根据这一假说,Upjohn公司在80年代开发了化合物U-50,488,但未见成功。δ受体的选择性激动剂开发,也有不少文献报道和专利,但未见临床成功的药物。因此,目前临床上的阿片类药物,主要是μ受体的激动剂。 前面提到的丁丙诺啡和扬子江的地佐辛,都是μ受体的部分激动剂。因此,这些药物的副作用有封顶效应,也就是达到一定程度后,不再随着剂量升高而增加,因此治疗窗口更大。而且,成瘾性也比吗啡等完全激动剂要小。丁丙诺啡的解离比较慢,而且是部分激动剂,因此可以用于抵抗完全激动剂的部分活性,临床上还用于治疗阿片类药物的急性戒断反应和戒毒。 临床上,具有双功能的镇痛药物,比如曲马多(tramadol)和他喷他多(tapentadol),也较为常见。这类药物虽然是较弱的μ受体激动剂,但同时抑制5-羟色胺 (serotonin) 和去甲肾上腺素 (norepinephrine) 等神经递质的重吸收,能够增加镇痛效果,特别是慢性神经痛。
如上所述,临床上已有许多阿片类药物,但效果和安全性仍然不理想。2006年,英国药理学杂志发表评论文章,回顾了75年的阿片研究,认为人类追求完美阿片药物的征程徒劳无功(5)。十年过去了,阿片受体的晶体结构被解析,作用机制的研究也取得巨大进步,基于各种策略的新药进入后期开发阶段。阿片药物的圣杯(Holy Grail)似乎近在眼前。
阿片类药物的当前研发策略
1.新剂型和防滥用制剂的开发
近年来,阿片类新实体分子并不多见,但新制剂和新剂量的505b(2)新药研发特别活跃,比如新的注射剂,透皮制剂,缓控释等。2月底,绿叶制药宣布CFDA批准丁丙诺啡透皮贴片的临床研究。这一产品由绿叶制药此前收购的子公司LuyePharma AG(绿叶制药德国)开发。BU08028虽然专利己过期,但开发有IP保护的新制剂仍然值得考虑。
另外,防药物滥用的制剂也是阿片类药物开发的热门方向。瘾君子为寻求最大快感,经常将处方药压碎后通过鼻腔吸入或注射给药。因此,防滥用的策略包括使用新辅料,使得片剂不容易被压碎,或者碎后形成胶体状物质不能注射。

Suboxone是丁丙诺啡和纳洛酮的复方口服药物,非常流行的阿片类镇痛药物,常年排在处方药的前50名。这个药物的防滥用策略比较有意思。前面提到,纳洛酮是阿片受体拮抗剂,可以抵消激动剂的药理活性,但这个药物有代谢的首过效应,如果口服,很快被肝脏代谢,将不会影响丁丙诺啡的镇痛效果。但如果瘾君子通过鼻腔吸入或注射给药,就会抵消丁丙诺啡的镇痛和产生快感的效果,因此达到防滥用的作用。代谢的首过效应,一向是口服药物开发头痛的问题,在这里却被巧妙利用,研发人员的创新精神令人赞叹。
2. 偏向性阿片受体配体 (Biased Opiate Receptor Ligand)
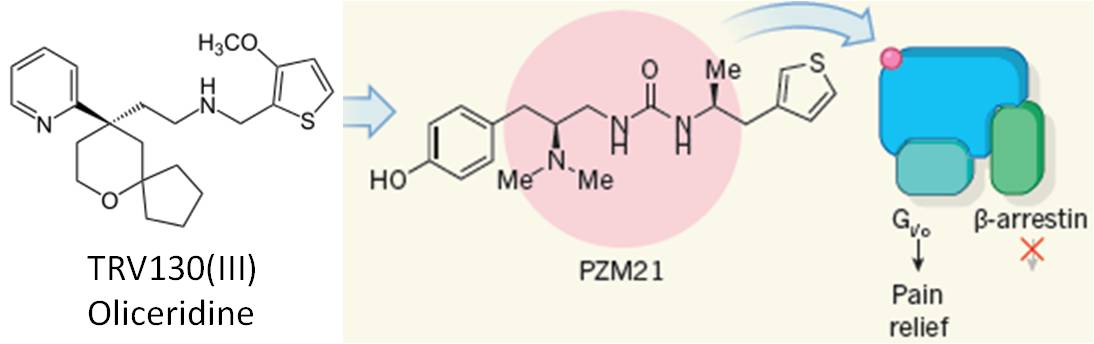
许多人对偏向性信号传导(biased signaling)这个概念不太了解。简单来说就是:一个受体,可以是多面手,通过不同的效应蛋白(effector),传导和调节下游的不同信号通路和功能。这是生物体系经济性的体现。因此,偏向性的配体,比如偏向性激动剂,它只特异性激活某种功能,而对该受体介导的其它功能没有激活作用。Roth等在1987年第一次描述了这种现象(6),随后,大量研究提到类似概念,比如功能选择性,差异化传导等等。1998年Jarpe等提出偏向性(biased)这个名词,逐渐被大家广泛接受(7)。 偏向性这个概念真正用于药物发现是最近的事。最接近成功的就是我们今天讨论的阿片类镇痛药物。前面提到,阿片类药物比如吗啡的作用主要是通过阿片μ受体传导的。μ受体是经典的GPCR,大量研究表明,μ受体镇痛作用是通过激活Gi信号,而便秘、抑制呼吸系统和成瘾性,更多地是通过下游β-arrestin通路调节。μ受体的偏向性激动剂,选择性激活Gi通路,而不影响β-arrestin信号通路。这就为开发理想的阿片药物提供了可能性。目前,进展最快的是Trevena的TRV130 (oliceridine)。2017年2月份,TRV130在两个三期临床中取得部分成功,手术后镇痛效果优于安慰剂,达到试验的一级终点。但在提高耐受性方面,却没有像人们预期地那样显示出优于吗啡的明显趋势。因此,偏向性激动剂这个假说,蒙上了一层阴影。Trevena在2月份宣布这一消息后,股票大跌40%。
其实,《Nature》2016年9月份发表了一篇文章(8),可以部分解释TRV130效果不佳的原因。这项研究的领导者正是解析GPCR晶体结构而获得2012年诺贝奖的美国斯坦福大学Brian Kobilka教授。基于阿片μ受体的晶体结构,通过合理设计和虚拟筛选,他们发现一个具有不同于吗啡骨架的μ受体偏向性激动剂PZM21。一系列动物试验表明,该化合物的偏向选择性高于TRV130,因此安全性指标明显优于TRV130。当然,这些优势需要临床确证,但从侧面提示,TRV130的选择性仍然不理想。其实,这也反映了偏向性药物开发的一个难点。因为不同功能的生物评价方法不一样,因此,在一定剂量下,很容易看到假的选择性。有时候,这种功能的选择性,可能只是来自评价方法的差异。
3. 双功能阿片受体激动剂
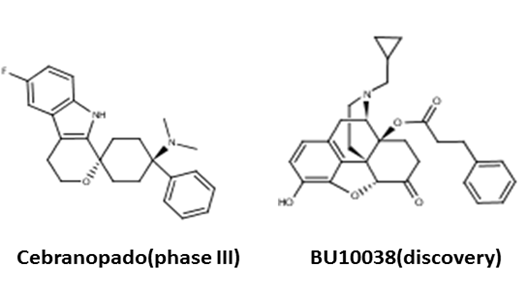
中枢神经系统药物,很少作用于单一靶点或受体,一般是多种作用机制的组合。阿片类药物也不例外。比如地佐辛,上海交通大学最近发表的研究表明,具有多种药理活性,包括阿片μ受体激动剂,κ受体的拮抗剂,以及去甲肾上腺素重吸收抑制剂(NRI)等(9)。 丁丙诺啡的成瘾性和副作用小于吗啡,有证据表明,部分原因是因为其具有弱的NOP受体激动作用。因此,一个值得探索的想法是,通过结构改造,保留丁丙诺啡的阿片μ受体的激动活性,同时增强其NOP受体活性,可能导致安全性更好的μ/NOP双功能药物。美国密执根大学的科学家于2004年首先报道了一个μ/NOP双功能化合物BU72(11)。同一个团队,又于2011年报道了文章开篇提到的丁丙诺啡类似物BU08028,其独特的药理活性,被认为来源于阿片μ受体和NOP受体双功能激动作用(12)。军科院的仲伯华教授极可能比他们更早发现了化合物BU08028。美国着名的民间研究机构之一,SRI International曾经与Astraea合作开发BU08028,但在一系列令人失望的动物实验后,停止了开发。 这里又带出新药研发过程中另外一个难点。那就是种属差异。BU08028在老鼠试验中,令人意外地,没有发现任何双功能的优势。在有的模型中,甚至镇痛活性还不如丁丙诺啡。这与最近的猴子试验形成鲜明对比:BU08028的镇痛活性是丁丙诺啡的10倍,而且瘙痒、呼吸系统抑制、成瘾性和身体依赖性等副作用非常小,全面优于丁丙诺啡。由于猴子更接近人类,这些效果也许能转移到临床试验中。这对于在研的μ/NOP双功能化合物应该是好消息,比如Grunenthal处于三期临床的Cebranopado,以及SRI开发的丁丙诺啡新类似物BU10038。
中枢神经系统药物,很少作用于单一靶点或受体,一般是多种作用机制的组合。阿片类药物也不例外。比如地佐辛,上海交通大学最近发表的研究表明,具有多种药理活性,包括阿片μ受体激动剂,κ受体的拮抗剂,以及去甲肾上腺素重吸收抑制剂(NRI)等(9)。 丁丙诺啡的成瘾性和副作用小于吗啡,有证据表明,部分原因是因为其具有弱的NOP受体激动作用。因此,一个值得探索的想法是,通过结构改造,保留丁丙诺啡的阿片μ受体的激动活性,同时增强其NOP受体活性,可能导致安全性更好的μ/NOP双功能药物。美国密执根大学的科学家于2004年首先报道了一个μ/NOP双功能化合物BU72(11)。同一个团队,又于2011年报道了文章开篇提到的丁丙诺啡类似物BU08028,其独特的药理活性,被认为来源于阿片μ受体和NOP受体双功能激动作用(12)。军科院的仲伯华教授极可能比他们更早发现了化合物BU08028。美国着名的民间研究机构之一,SRI International曾经与Astraea合作开发BU08028,但在一系列令人失望的动物实验后,停止了开发。 这里又带出新药研发过程中另外一个难点。那就是种属差异。BU08028在老鼠试验中,令人意外地,没有发现任何双功能的优势。在有的模型中,甚至镇痛活性还不如丁丙诺啡。这与最近的猴子试验形成鲜明对比:BU08028的镇痛活性是丁丙诺啡的10倍,而且瘙痒、呼吸系统抑制、成瘾性和身体依赖性等副作用非常小,全面优于丁丙诺啡。由于猴子更接近人类,这些效果也许能转移到临床试验中。这对于在研的μ/NOP双功能化合物应该是好消息,比如Grunenthal处于三期临床的Cebranopado,以及SRI开发的丁丙诺啡新类似物BU10038。
个人的一些思考
1.药理评价和生物基础研究是中国新药研发的短板
BU08028是极具前景的新型镇痛候选药物。中国军科院和美国密执根大学的科学家相继独立发现了该化合物,却因为没有合适的生物评价方法,而不能发现其潜力,最终被放弃。充分说明,药理评价是新药研发的“眼睛”,缺少这只眼睛,就是重磅炸弹药物摆在你的面前,你也看不到。而生物基础研究是建立合理药理评价的前提。最近几年,中国新药研发飞速发展,无论是政策环境和技术实力都有长足的进步,然而,药理评价和生物基础研究始终是制约我们新药研发的瓶颈,特别是对于原创新药的研发,尤其如此。 中药现代化,在青蒿素获得诺贝尔奖后,又得到政府的空前重视。这次十三五重大专项,就专门有一个青蒿素的专项。从中药或天然产物中寻找先导化合物是不是中药的发展方向,我们暂且不论。但没有以生物研究为基础的创新筛选方法,要取得成功是非常困难的。多年来,我们天然产物药物的突破性发现不多,并不是我们的分离技术、结构鉴定有差距,而是大量分离的化合物以简单的细胞毒筛选草草收场。我们说中药是一个宝库,其实化学合成也是一个宝库。每年合成那么多的化合物,市场上也有大量商业化合物库,很难说,其中就没有类似BU08028的金子,只是我们没有眼睛去发现这些金子。因此,中药现代化和化学合成药物其实是面临同样的问题。 在这个案例中,还涉及一个争论,以化学结构的类似性来判断药物的创新性。BU08028再次证明,化学结构的微小变化,可能带来药理活性天翻地覆的变化。据说,结构太类似,也是军科院当年没有选择ADP2进一步开发的原因之一,十分令人遗憾。
2.新药研发要瞄准真正的临床需求
地佐辛的成功,当然和扬子江强大的销售渠道有关。独家,注射剂,高定价,科室广泛等也有中国式重磅炸弹的特征。但地佐辛和充斥中国市场的“神药”还是有所不同。因为它确实满足了临床的刚性需求。经常有人问我,立项时如何考虑治疗领域。有一个简单的办法,那就是对比中国和成熟市场,发现差距。据IMS的一个数据,镇痛药物在美国的药占比为6.4%,而中国市场只有1.2%,差距明显,发展空间巨大。美国十分重视肿瘤等绝症病人的临终关怀。在病人的最后日子里,大量使用吗啡等控制药品,减少病人的痛苦。阿片类药物的使用和管理水平非常高。而在中国和印度等大国,成瘾性镇痛药物被严格控制。美国人均吗啡的用量高达75mg以上,而印度人均不足1mg。中国没有准确的数据,估计为0.01mg左右。2016年7月,《Nature》上以“另类阿片问题”为题,发表一篇评论文章,呼吁增加不发达国家阿片药物的可及性(12)。 中国对吗啡的管控十分严格。而地佐辛的独特药理特征(部分及混合激动-拮抗剂)使得其成瘾性和身体依赖小于吗啡,在中国仅作为精神二类物质管制。另外,由于吗啡长期受到严格控制,中国镇痛和麻醉的管理水平不够高,而这些药物的副作用有时是致死的,在当今的医患矛盾下,医生使用更加谨慎。而前面提到,地佐辛是μ受体的部分激动剂,因此,抑制呼吸系统等副作用有封顶效应,不易因为过量而致死,这是地佐辛受到中国医生欢迎的重要原因。看到扬子江的一个调研数据,医生对地佐辛好评比例明显高于吗啡,但病人的评价却相当。药品是个特殊商品,医生才是真正的消费者。因此,我们要明白,从商业的角度上,医生的需求才是真正的临床需求,有时候,并不完全与病人的需求一致。我国以药养医带来的困境也有类似的因素。当前的改革方向,就是要理顺医生和病人的需求,力求一致,缓解医患矛盾。 作为研发人员,我们当然还是要首先考虑病人的需求。当我们眼里有了病人的需求,就能产生创新的想法。比如,激酶抑制剂的研发是国内的热门方向。除了常规的提高疗效、克服耐药性等想法,有没有人去想,这是给癌症病人使用的药物,而癌痛是他们最常见的痛苦。最近的研究显示,诱导PDGFR的磷酸化,下调μ受体,是阿片类药物产生耐受性的重要原因。而神药伊马替尼可以抑制PDGFR的磷酸化,从而缓解疼痛,增敏吗啡的功效(13,14)。可以推测,有PDGFR激酶活性的替尼,都有类似的功效。难怪有的病人服用替尼药物后,虽然肿瘤的疗效不佳,但疼痛得到缓解。显然,在做激酶抑制剂选择性的时候,可以重新考虑PDGFR的活性。没有作过细致的调研,这种想法也许不成熟。只是想说明,考虑临床需求,有许多角度,创新可以是任何地方。前面提到的利用首过效应防药物滥用,正是这一观点的例证。
结语
类似军科院和扬子江的阿片镇痛药物的故事还有很多,都值得去总结。从前人失败和成功的经历中可以学到很多东西。最终的目的还是要用于指导今后的新药研发。有些信息和评论可能不准确或者考虑不周,还请读者批评指正。
类似军科院和扬子江的阿片镇痛药物的故事还有很多,都值得去总结。从前人失败和成功的经历中可以学到很多东西。最终的目的还是要用于指导今后的新药研发。有些信息和评论可能不准确或者考虑不周,还请读者批评指正。
参考文献
A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. PNAS 2016, 113(37),E5511-8
Buprenorphine analogue BU08028 is one step closer to the Holy Grail of opioid research. PNAS 2016, 113(37), 10225-7
丁丙诺啡类似物的合成。吴波硕士论文
A brief history of opiates, opioid peptides, and opioid receptors. PNAS 1993, 90, 5391-5393.
75 years of opioid research: the exciting butvain quest for the Holy Grail。British Journal of Pharmacology (2006) 147, S153–S162
Multiple mechanisms of serotonergic signal transduction. Life Sci 1987,41:1051–1064
Substance P acts as a biased agonist toward neuropeptide and chemokine receptors. J Biol Chem 1998, 273:3097–3104
Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016 Sep8;537(7619):185-190
Dezocine exhibits antihypersensitivity activities in neuropathy through spinal μ-opioid receptor activation and norepinephrine reuptake inhibition. Sci Rep 2017 Feb23;7:43137
Characterization of the complex morphinan derivative BU72 as a high efficacy, long-lasting mu-opioid receptor agonist. Eur J Pharmacol. 2004 Sep 19;499(1-2):107-16
The first universal opioid ligand BU08028:characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J Pharmacol Exp Ther. 2011Mar;336(3):952-61
The other opioid issue. Nature 2016,535, s16-17
Wang Y, Barker K, Shi S,Diaz M, Mo B, Gutstein HB (2012) Blockade of PDGFR-activation eliminates morphine analgesic tolerance. Nat Med 2012 , 18:385–387
Pain and Poppies: The Good, the Bad, and the Ugly of Opioid Analgesics. The Journal of Neuroscience 2015 , 35(41):13879 –13888
[责任编辑]杜新忠